Drug side effects inevitable since basic physics enabled early biochemistry, says study
25 May 2013
A new study of both computer-created and natural proteins suggests that the number of unique pockets – sites where small molecule pharmaceutical compounds can bind to proteins – is surprisingly small, meaning drug side effects may be impossible to avoid. The study also found that the fundamental biochemical processes needed for life could have been enabled by the simple physics of protein folding.
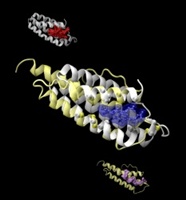 |
| Image shows a naturally heme-bound (blue) protein structure (white, pocket in red), superimposed onto an artificial protein structure (yellow, pocket in purple) that has both a similar protein fold and a similar pocket. The pocket-like features of proteins can be artificially generated in stable proteins without invoking evolutionary selection. (Image courtesy of Mu Gao) |
Studying a set of artificial proteins and comparing them to natural proteins, researchers at the Georgia Institute of Technology have concluded that there may be no more than about 500 unique protein pocket configurations that serve as binding sites for small molecule ligands. Therefore, the likelihood that a molecule intended for one protein target will also bind with an unintended target is significant, said Jeffrey Skolnick, a professor in the School of Biology at Georgia Tech.
''Our study provides a rationalisation for why a lot of drugs have significant side effects – because that is intrinsic to the process,'' said Skolnick. ''There are only a relatively small number of different ligand binding pockets. The likelihood of having geometry in an amino acid composition that will bind the same ligand turns out to be much higher than anyone would have anticipated. This means that the idea that a small molecule could have just one protein target can't be supported.''
Research on the binding pockets was published May 20 in the early edition of the journal Proceedings of the National Academy of Sciences. The research was supported by the National Institutes of Health (NIH).
Skolnick and collaborator Mu Gao have been studying the effects of physics on the activity of protein binding, and contrasting the original conditions created by the folding of amino acid residues against the role played by evolution in optimizing the process.
''The basic physics of the system provides the mechanism for molecules to bind to proteins,'' said Skolnick, who is director of the Center for the Study of Systems Biology at Georgia Tech. ''You don't need evolution to have a system that works on at least a low level. In other words, proteins are inherently capable of engaging in biochemical function without evolution's selection. Beyond unintended drug effects, this has a lot of implications for the biochemical component of the origins of life.''
Binding pockets on proteins are formed by the underlying secondary structure of the amino acids, which is directed by hydrogen bonding in the chemistry. That allows formation of similar pockets on many different proteins, even those that are not directly related to one another.
''You could have the same or very similar pockets on the same protein, the same pockets on similar proteins, and the same pockets on completely dissimilar proteins that have no evolutionary relationship. In proteins that are related evolutionarily or that have similar structures, you could have very dissimilar pockets,'' said Skolnick, who is also a Georgia Research Alliance Eminent Scholar. ''This helps explain why we see unintended effects of drugs, and opens up a new paradigm for how one has to think about discovering drugs.''
The implications of this ''biochemical noise'' for the drug discovery process could be significant. To counter the impact of unintended effects, drug developers will need to know more about the available pockets so they can avoid affecting binding locations that are also located on proteins critical to life processes. If the inevitable unintended binding takes place on less critical proteins, the side effects may be less severe.
In addition, drug development could also move to a higher level, examining the switches that modulate the activity of proteins beyond binding sites. That may require a different approach to drug development.
''The strategy for minimising side effects and maximising positive effects may have to operate at a higher level,'' Skolnick said. ''You are never going to be able to design unintended binding effects away. But you can minimise the undesirable effects to some extent.''
In their study, Skolnick and Gao used computer simulations to produce a series of artificial proteins that were folded according the laws of physics, but not optimised for function. Using an algorithm that compares pairs of pockets and assesses the statistical significance of their structural overlap, they analysed the similarity between the binding pockets in the artificial proteins and the pockets on a series of native proteins. The artificial pockets all had corresponding pockets on the natural proteins, suggesting that the simple physics of folding has been a major factor in development of the pockets.
''This is how life, at least the biochemistry of life, could have gotten started,'' said Skolnick. ''Evolution would have optimised the functions, but you don't need that to get started at a low level of efficiency. If you had a soup of our artificial proteins, even with no selection you could at least do low-level biochemistry.''
Though the basic biochemistry of life was made possible by simple physics, optimising the binding process to allow the efficiencies seen in modern organisms would have required evolutionary selection.
''This is the first time that it has been shown that side effects of drugs are an inherent, fundamental property of proteins rather than a property that can be controlled for in the design,'' Skolnick added. ''The physics involved is more important than had been generally appreciated.
Research reported in this news release was supported by the Institute of General Medical Sciences of the National Institutes of Health.


















